AWSEM - Associative Memory, Water Mediated, Structure and Energy Model
Welcome to the webpage of AWSEM-MD, a coarse-grained protein simulation package that is being developed and maintained by the Papoian lab at the University of Maryland and the Wolynes lab at Rice University. This webpage contains documentation and examples for the AWSEM software package, which is implemented as a package for the LAMMPS Molecular Dynamics Program. The source code for this package is downloadable for scientific use.
Background
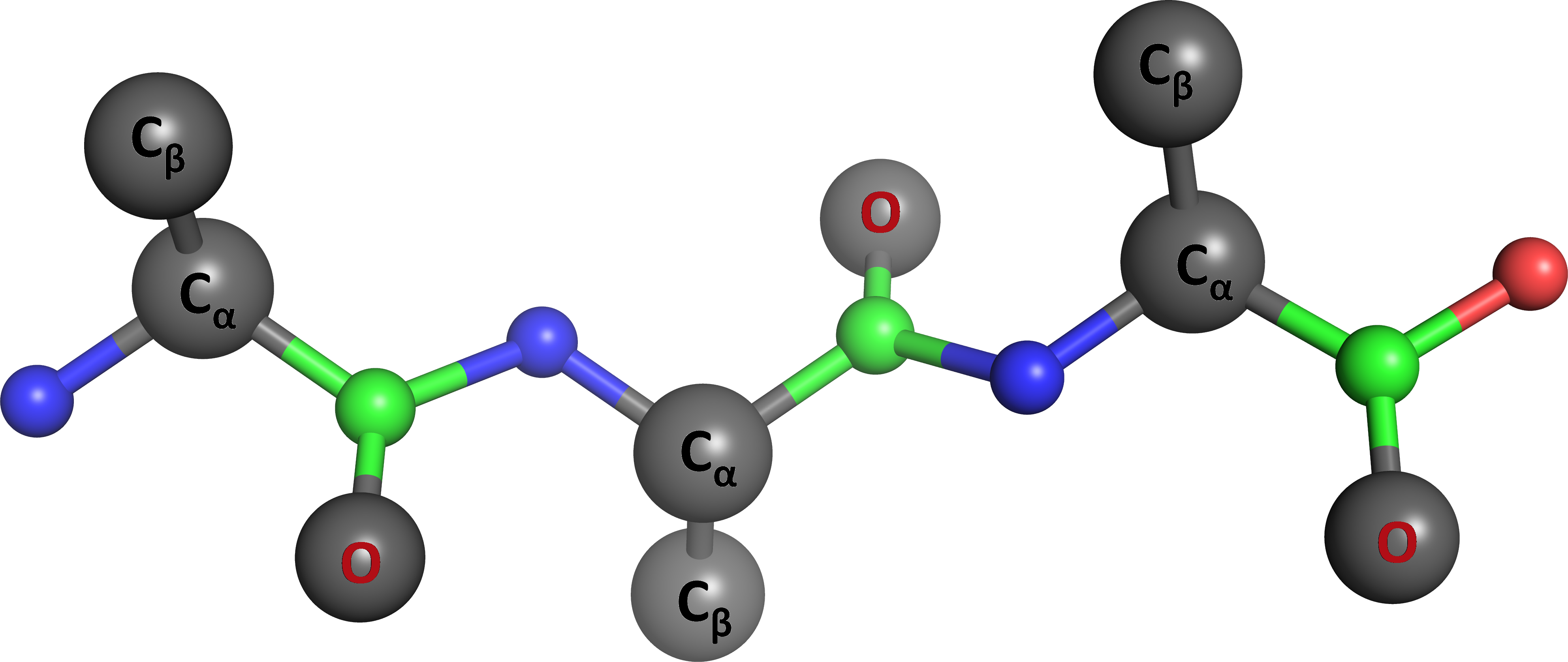
Proteins and other biomolecules can be simulated on a computer primarily in two ways: relying either on atomistic or coarse-grained force fields. The latter approaches, which enable treating large systems at long timescales, come in with rather broad variety of choices, compared with the atomistic models. In particular, many useful coarse-grained protein force fields require prior knowledge of the native structure for the specific protein target, often with the stated goal of simulating protein folding kinetics and dynamics. These methods, however, cannot be used when the target protein structure is unknown or non-native interactions play a significant role. To address such problems, several coarse-grained protein force fields have been developed in the last 30 years or so that allow de novo structure prediction even in the absence of sequence homology. The AWSEM potential is a prominent member of this latter group, where various research groups have successfully demonstrated its broad applications in monomeric protein structure prediction, binding predictions of dimers and multimeric assemblies, folding of membrane proteins, and structural and kinetic studies of protein-DNA complexes. AWSEM naturally covers the whole spectrum between native-structure-based and de novo methods, and can be used both with the knowledge of native structures, and without such explicit knowledge. In the latter case it relies solely on sequence information. Because AWSEM's potential is analytical and differentiable, it is currently implemented as a molecular dynamics algorithm (AWSEM-MD).
The AWSEM force field grew out of the Associative Memory Hamiltonian (AMH) family of protein modeling potentials, developed over many years by Professor Peter Wolynes and his group at the University of Illinois at Urbana-Champaign, the University of California in San Diego and Rice University. The contact and water-mediated interactions were developed by Papoian, Ulander, and Wolynes in 2004 ("Water in Protein Structure Prediction" ; Proc. Natl. Acad. Sci. USA; 2004, 101 , 3352), when the revised AMH was renamed AMW (where W stands for water). The original AMH/AMW programs were written in FORTRAN by many people over 20 years, including Mark Friedrichs, Richard Goldstein, Zaida Luthey-Schulten, Kristin Koretke, Corey Hardin, Michael Eastwood, Michael Prentiss, Garegin Papoian, Johan Ulander, Chenghang Zong, Cecillia Clementi and Vanessa Oklejas, among others.
The AWSEM potential is based on a three-bead per amino-acid residue representation of protein chains, having many terms representing the backbone stereochemistry, independent and cooperative hydrogen bonding, water-mediated tertiary interactions, and biasing local structural preferences based on short fragment memories. Dr. Aram Davtyan, while being a graduate student in Prof. Garegin Papoian's group at the University of Maryland, designed the architecture of AWSEM's code, and implemented its main features in C++, as an add-on package compatible with the LAMMPS simulation platform. Dr. Nicholas Schafer and Dr. Weihua Zheng from Prof. Peter Wolynes' group at Rice University also made significant early contributions to AWSEM's development, applications and documentation. Currently, AWSEM software is being actively developed by many members and alumni of the Papoian and Wolynes laboratories, as well as other scientists. Dr. Aram Davtyan continues to serve as software's lead developer.
If you use the program, you agree to cite the following publication that introduced AWSEM and contains a complete description of the forcefield:
Aram Davtyan, Nicholas P. Schafer, Weihua Zheng, Cecilia Clementi, Peter G. Wolynes, and Garegin A. Papoian, "AWSEM-MD: Protein Structure Prediction Using Coarse-Grained Physical Potentials and Bioinformatically Based Local Structure Biasing", J. Phys. Chem. B 2012 116 (29), 8494-8503
Funding sources

